TL;DR: NAD+ is a central coenzyme in cellular energy metabolism that declines with age. This decline disrupts sirtuin signaling (SIRT1/SIRT3), impairs mitochondrial function, and activates competing PARP-mediated pathways. NAD+ longevity research now focuses on precursor molecules – NMN and NR – as tools for investigating age-related metabolic deterioration in preclinical models.
Table of Contents
- What Is NAD+ and Why Does It Matter in Longevity Research?
- How Is NAD+ Synthesized in Mammalian Cells?
- What Roles Do SIRT1 and SIRT3 Play in NAD+ Longevity Research?
- How Does PARP Competition Contribute to NAD+ Decline?
- NMN vs NR – How Do NAD+ Precursors Compare in Research Models?
- What Do Preclinical Models Reveal About NAD+ and Hallmarks of Aging?
- Frequently Asked Questions
What Is NAD+ and Why Does It Matter in Longevity Research?
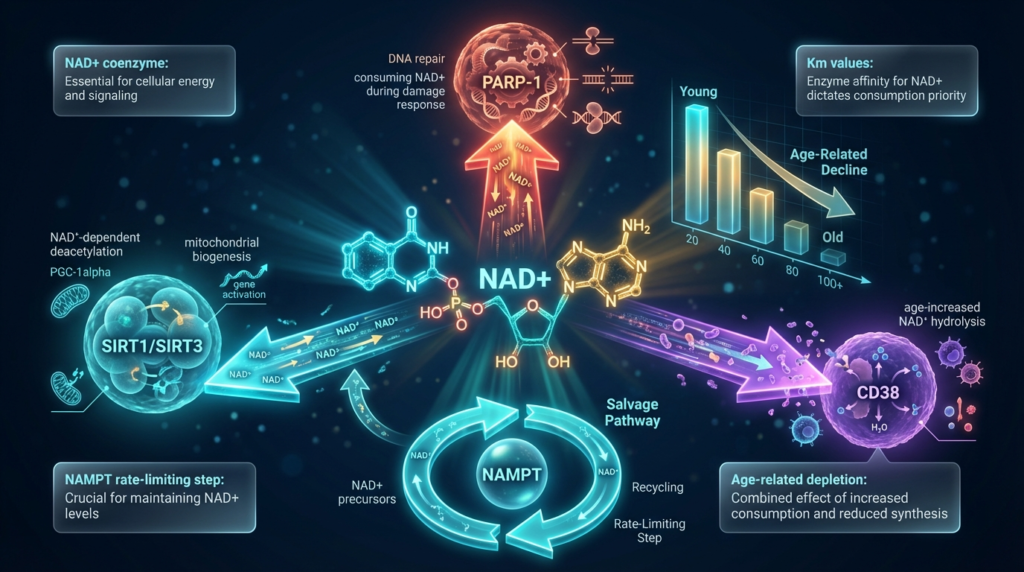
Nicotinamide adenine dinucleotide (NAD+) is a coenzyme present in every living cell, functioning as an essential electron carrier in redox reactions and as a co-substrate for NAD+-consuming enzymes. NAD+ longevity research has expanded rapidly over the past decade because cellular NAD+ concentrations decline measurably during the aging process, and this decline correlates with functional deterioration across multiple organ systems. [1]
In its oxidized form, NAD+ accepts electrons during glycolysis and the citric acid cycle, transferring them to the mitochondrial electron transport chain (ETC) where ATP is generated. Beyond this classical metabolic role, NAD+ serves as a required co-substrate for three major enzyme families: sirtuins (SIRT1-7), poly-ADP-ribose polymerases (PARPs), and CD38/CD157 ectoenzymes. Each of these enzymes consumes NAD+ during catalysis, cleaving it into nicotinamide (Nam) and an ADP-ribose moiety. [1]
The significance of NAD+ in aging biology stems from this dual role. As NAD+ levels fall, the enzymes that depend on it – particularly the sirtuins – lose catalytic capacity. Researchers have observed that restoring NAD+ levels in aged preclinical models can reverse certain aspects of mitochondrial dysfunction, re-establishing metabolic function to levels observed in younger organisms. [2]
How Is NAD+ Synthesized in Mammalian Cells?
Mammalian cells produce NAD+ through three principal biosynthetic routes:
- The de novo pathway – converts the amino acid tryptophan through a multi-step process involving quinolinic acid phosphoribosyltransferase (QPRT), ultimately generating NAD+ via nicotinic acid mononucleotide (NaMN)
- The Preiss-Handler pathway – salvages nicotinic acid (NA, a form of vitamin B3) through nicotinic acid phosphoribosyltransferase (NAPRT), producing NaMN as an intermediate
- The salvage pathway – recycles nicotinamide (Nam) through nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme in mammalian NAD+ biosynthesis
The salvage pathway is quantitatively dominant in most mammalian tissues. NAMPT converts Nam to nicotinamide mononucleotide (NMN), which is then adenylylated by nicotinamide mononucleotide adenylyltransferases (NMNATs) to form NAD+. This pathway is particularly significant because every NAD+-consuming reaction produces Nam as a byproduct, creating a recycling loop. [1]
Two additional precursor entry points have attracted research attention:
- Nicotinamide riboside (NR) enters the pathway through nicotinamide riboside kinases (NRK1/NRK2), which phosphorylate NR directly to NMN
- Nicotinamide mononucleotide (NMN) can be taken up by cells and converted to NAD+ in a single adenylylation step
NAMPT expression itself is regulated by the circadian clock via the BMAL1/CLOCK transcriptional complex, linking NAD+ biosynthesis to circadian rhythm. SIRT1 in turn modulates BMAL1/CLOCK activity, creating a feedback loop that couples NAD+ production to time-of-day metabolic demands. [1]
What Roles Do SIRT1 and SIRT3 Play in NAD+ Longevity Research?
SIRT1 and SIRT3 are NAD+-dependent deacetylases that regulate distinct but complementary aspects of cellular metabolism, making them central targets in NAD+ longevity research.
SIRT1 – Nuclear and Cytoplasmic Regulation
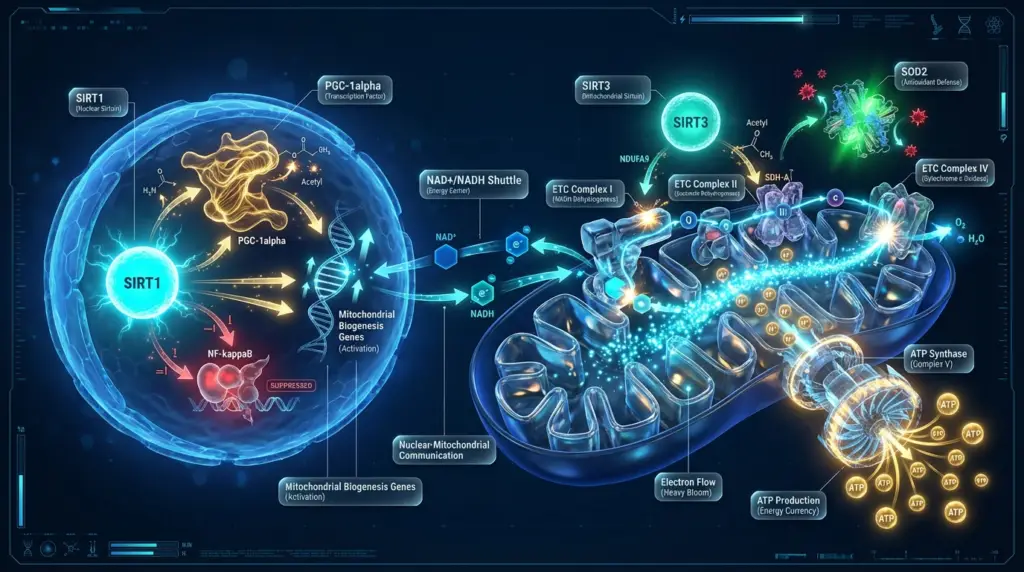
SIRT1 is the most extensively studied mammalian sirtuin. It deacetylates a broad range of substrates including:
- PGC-1alpha – a master regulator of mitochondrial biogenesis
- FOXO transcription factors – involved in stress resistance and metabolic adaptation
- p53 – a key mediator of cellular senescence and DNA damage response
- NF-kappaB – a central regulator of inflammatory signaling
When NAD+ levels decline during aging, SIRT1 activity decreases proportionally. Research by Gomes et al. demonstrated that this decline in nuclear NAD+ produces a “pseudohypoxic” state – an accumulation of HIF-1alpha under normoxic conditions – that disrupts nuclear-mitochondrial communication and selectively reduces mitochondrial-encoded OXPHOS subunit expression. [2]
SIRT3 – The Mitochondrial Sentinel
SIRT3 resides in the mitochondrial matrix, where it deacetylates components of the electron transport chain (ETC) and the citric acid cycle. Key SIRT3 substrates include:
- Complex I subunits (NDUFA9) – critical for electron entry into the ETC
- Complex II subunit (SDH-A) – linking the TCA cycle to OXPHOS
- SOD2 (superoxide dismutase 2) – the primary mitochondrial antioxidant enzyme
- Acetyl-CoA synthetase 2 (AceCS2) – regulating mitochondrial acetyl-CoA metabolism
Together, SIRT1 and SIRT3 form a coordinated regulatory network. SIRT1 drives the transcriptional programs that produce new mitochondrial components from the nucleus, while SIRT3 maintains functional efficiency of the existing mitochondrial machinery. When NAD+ becomes limiting, both arms of this network weaken simultaneously.
How Does PARP Competition Contribute to NAD+ Decline?
PARP-1 (poly-ADP-ribose polymerase 1) is the single largest consumer of NAD+ in the nucleus during DNA damage response. PARP-1 detects single-strand DNA breaks and catalyzes the addition of poly-ADP-ribose (PAR) chains to target proteins, consuming one molecule of NAD+ per ADP-ribose unit added. A single activated PARP-1 enzyme can deplete local NAD+ pools within minutes. [3]
The PARP-sirtuin competition model explains a core mechanism of age-related NAD+ depletion:
- DNA damage accumulates with age from oxidative stress, replication errors, and environmental exposures
- PARP-1 activation increases to repair this mounting damage, consuming progressively more NAD+
- NAD+ availability for sirtuins decreases, reducing SIRT1/SIRT3 deacetylase activity
- Mitochondrial function declines as SIRT1 can no longer adequately activate PGC-1alpha or maintain nuclear-mitochondrial communication
- Increased mitochondrial dysfunction generates more reactive oxygen species (ROS), causing further DNA damage – creating a self-reinforcing cycle
A third NAD+ consumer, CD38, also increases in expression during aging. CD38 is an ectoenzyme that hydrolyzes NAD+ to produce cyclic ADP-ribose and Nam. Research indicates that CD38 expression rises significantly in aged tissues and may account for a substantial portion of age-related NAD+ decline, independent of PARP activation. [7]
The combined drain from PARP-1, CD38, and baseline sirtuin activity exceeds the biosynthetic capacity of the NAMPT-mediated salvage pathway in aged tissues, resulting in the net NAD+ deficit observed in aging research models.
NMN vs NR – How Do NAD+ Precursors Compare in Research Models?
Two NAD+ precursor molecules – nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR) – have emerged as the primary investigational compounds for restoring NAD+ levels in preclinical research. Both feed into the salvage pathway but differ in their biochemistry, pharmacokinetics, and documented research outcomes.
| Parameter | NMN (Nicotinamide Mononucleotide) | NR (Nicotinamide Riboside) |
|---|---|---|
| Molecular weight | 334.2 Da | 255.2 Da |
| Entry enzyme | Converted directly by NMNATs to NAD+ | Phosphorylated by NRK1/NRK2 to NMN |
| Steps to NAD+ | 1 (adenylylation) | 2 (phosphorylation, then adenylylation) |
| Cellular uptake | Via Slc12a8 transporter (tissue-specific) | Via equilibrative nucleoside transporters |
| Key preclinical findings | Reversed age-associated metabolic dysfunction in mice [6] | Improved mitochondrial and stem cell function, extended mouse lifespan [4] |
| Oral bioavailability | Demonstrated in murine models [6] | Demonstrated in mice and human pharmacokinetic study [5] |
| NAAD formation | Observed as intermediate | Confirmed as sensitive biomarker of NAD+ repletion [5] |
| Circadian sensitivity | NAMPT-dependent recycling is clock-regulated | NRK pathway is less circadian-dependent |
| Stability | Less stable in aqueous solution at room temperature | More stable in powder and solution form |
| Research stage | Extensive preclinical, emerging clinical | Extensive preclinical, multiple human trials completed |
Yoshino et al. demonstrated that NMN administration to aged mice reversed metabolic dysfunction associated with diet- and age-induced models, restoring NAD+ levels and improving metabolic parameters. [6] Zhang et al. showed that NR supplementation rejuvenated muscle stem cells in aged mice, prevented stem cell senescence, and extended lifespan through a mitochondrial unfolded protein response (UPR-mt) mechanism. [4]
Trammell et al. established the first human pharmacokinetic profile of NR, demonstrating dose-dependent increases in blood NAD+ metabolites and identifying NAAD (nicotinic acid adenine dinucleotide) as a highly sensitive biomarker of effective NAD+ repletion. [5]
Researchers studying NAD+ longevity research pathways can access these compounds for qualified in vitro investigation. Molecular Edge Peptides supplies research-grade biofermented NAD+ (1000mg) manufactured to >99% purity with full Certificate of Analysis documentation for qualified laboratory use.
What Do Preclinical Models Reveal About NAD+ and Hallmarks of Aging?
NAD+ decline intersects with multiple recognized hallmarks of aging, making it a convergence point for several age-related deterioration pathways:
Mitochondrial Dysfunction
The most directly documented consequence. Declining NAD+ reduces ETC electron flow by limiting NADH availability for Complex I, while simultaneously reducing SIRT3-mediated deacetylation of ETC components. Gomes et al. demonstrated that raising NAD+ levels in aged mice restored mitochondrial function to levels characteristic of young animals in a SIRT1-dependent manner. [2]
Genomic Instability
PARP-1 requires NAD+ for DNA repair. As basal NAD+ drops, the efficiency of DNA single-strand break repair decreases, contributing to mutation accumulation.
Cellular Senescence
NAD+ depletion in stem cell compartments accelerates entry into senescence. Zhang et al. showed that NR supplementation delayed senescence in neural stem cells, melanocyte stem cells, and muscle stem cells in aged mice. [4]
Altered Intercellular Communication
NAD+-dependent CD38 activity on cell surfaces modulates extracellular NAD+ availability, affecting paracrine signaling between cells.
Deregulated Nutrient Sensing
The SIRT1-AMPK-mTOR axis – a central nutrient-sensing network – requires adequate NAD+ for SIRT1 function. NAD+ decline uncouples this sensing mechanism from actual nutrient status.
Research into telomere biology and NAD+ also connects to broader longevity science. The peptide Epithalon has been investigated in telomerase activation contexts, representing a complementary area of aging research at the cellular level. The mitochondria-derived peptide MOTS-c is another compound studied for its role in mitochondrial-nuclear communication and metabolic regulation.
For researchers exploring the broader landscape of aging biology protocols, the 2026 Master Index of Peptide Research Protocols, Hubs, and Standards provides a comprehensive directory of research resources and methodological frameworks.
Frequently Asked Questions
What is NAD+ and why is it studied in longevity research?
NAD+ (nicotinamide adenine dinucleotide) is a coenzyme required for cellular energy production and as a co-substrate for enzymes including sirtuins and PARPs. NAD+ longevity research focuses on the observation that cellular NAD+ concentrations decline measurably with age across tissues, and this decline correlates with mitochondrial dysfunction, impaired DNA repair, and reduced sirtuin activity in preclinical models. Restoring NAD+ through precursor supplementation has shown promise in reversing age-related metabolic deterioration in laboratory settings.
How do sirtuins depend on NAD+ for their function?
Sirtuins (SIRT1-7) are a family of NAD+-dependent deacetylases and ADP-ribosyltransferases. They require NAD+ as a co-substrate to remove acetyl groups from target proteins, consuming one NAD+ molecule per deacetylation reaction. When cellular NAD+ levels fall below the Km (Michaelis constant) of a given sirtuin, its catalytic activity decreases proportionally. SIRT1 and SIRT3 are the most studied in aging contexts, regulating nuclear gene expression and mitochondrial protein function respectively.
What is the difference between NMN and NR as NAD+ precursors?
NMN (nicotinamide mononucleotide) and NR (nicotinamide riboside) are both NAD+ precursor molecules that enter the salvage biosynthetic pathway at different points. NMN requires a single adenylylation step to become NAD+, while NR must first be phosphorylated to NMN by NRK1/NRK2 kinases before adenylylation. NR has demonstrated oral bioavailability in both mice and humans through clinical pharmacokinetic studies, while NMN uptake involves tissue-specific transporter proteins.
Why does NAD+ decline with aging?
NAD+ decline during aging results from increased consumption by DNA repair enzymes (PARP-1), elevated CD38 ectoenzyme expression, and decreased activity of the rate-limiting biosynthetic enzyme NAMPT. DNA damage accumulates over time, activating PARP-1 and consuming more NAD+. CD38 expression increases in aged tissues, further depleting available NAD+. Simultaneously, NAMPT expression decreases in aged tissues, reducing the rate of NAD+ recycling from nicotinamide.
How does PARP-1 compete with sirtuins for NAD+?
PARP-1 and sirtuins both consume NAD+ as a co-substrate but serve different cellular functions. PARP-1 uses NAD+ for DNA repair by attaching poly-ADP-ribose chains to proteins near DNA break sites. Sirtuins use NAD+ for protein deacetylation and metabolic regulation. Because PARP-1 has a lower Km for NAD+ and higher catalytic consumption rate, it effectively outcompetes sirtuins when DNA damage increases, redirecting the limited NAD+ pool toward repair at the expense of metabolic regulation.
What role does mitochondrial function play in NAD+ aging research?
Mitochondrial function is central to NAD+ longevity research because the ETC depends on NADH (reduced NAD+) to generate ATP through oxidative phosphorylation. Age-related NAD+ decline reduces SIRT3-mediated maintenance of ETC complex integrity, decreases SIRT1-driven mitochondrial biogenesis via PGC-1alpha, and impairs the nuclear-mitochondrial communication required for coordinated OXPHOS subunit expression. Published preclinical data demonstrate that NAD+ repletion can restore mitochondrial function parameters in aged organisms.
Disclaimer: All products sold by Molecular Edge Peptides are strictly intended for laboratory research use only (in vitro). They are not approved for human or animal consumption, or for any form of therapeutic, diagnostic, or clinical use. The information in this article is for educational and scientific reference purposes only. We do not provide usage instructions, dosing guidelines, or any advice regarding personal application of our products. Always consult relevant regulatory frameworks before conducting research with these compounds.
References
- Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24(8):464-471. PubMed
- Gomes AP, Price NL, Ling AJ, et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155(7):1624-1638. PubMed
- Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015;350(6265):1208-1213. PubMed
- Zhang H, Ryu D, Wu Y, et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352(6292):1436-1443. PubMed
- Trammell SA, Schmidt MS, Weidemann BJ, et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016;7:12948. PubMed
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14(4):528-536. PubMed
- Xie N, Zhang L, Gao W, et al. NAD+ deficiency is a common central pathological factor of a number of diseases and aging: mechanisms and therapeutic implications. Antioxid Redox Signal. 2020;32(13):894-916. PubMed
For in vitro research use only. Not for human or animal consumption.
